
24일 업계에 따르면 SK바이오팜의 뇌전증 치료제 엑스코프리는 올 1분기 미국에서 317억원의 매출을 올렸다. 지난해 매출(782억원)의 40% 가량을 1분기에 냈다. 회사는 올해 매출 목표를 1600억원으로 잡았다.
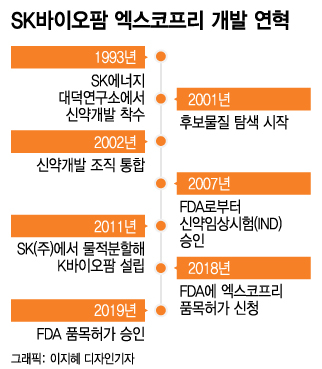
전 세계에 진출해 K-신약의 대표주자로 꼽히는 엑스코프리에 대한 연구는 지난 2001년으로 거슬러 올라간다. 이때부터 연구진은 신약 후보물질을 탐색하기 시작했다. 2000개 이상의 화합물을 합성한 후에야 엑스코프리를 발굴했다.
국내 업계에서는 현지 파트너사를 통해 임상시험을 진행하는 것이 시간과 비용 측면에서 효율적이라고 평가한다. 임상 단계에서 후보 물질을 글로벌 제약사에 기술수출 하는 것을 이상적인 사업 모델로 꼽는 이유다. FDA가 임상시험 기준으로 보는 환자 관리, 임상시험 기관 관리 기준 등이 국내 식품의약품안전처와는 다르기 때문이다. 필요한 서류도 차이가 있다. 식약처 기준에 따라 진행해온 연구진이 이 기준을 이해하고 맞추기까지는 시간이 걸릴 수밖에 없다. 비용도 문제다. 특히 임상 3상은 1000여명이 넘는 환자들을 대상으로 진행하기 때문에 관리 비용이 이전 단계에 비해 대폭 늘어난다.
업계 관계자는 "임상시험 승인을 받은 후 진행하는 과정에서 환자, 임상 기관, 데이터 관리 등 세부적인 절차가 식약처와 FDA의 기준이 다른 것으로 안다"라며 "이 때문에 국내 기업이 파트너사 없이 직접 현지 임상을 진행하면 연구 인력이 애를 먹는 것으로 알려져 있다"라고 했다.
이 시각 인기 뉴스
임상시험을 마친 후 FDA에 품목허가 신청을 위해 필요한 데이터를 도출하는 것도 쉽지 않았다. SK바이오팜은 당시 230여만 페이지의 자료를 작성했다.
업계에서는 신약 개발을 흔히 '낙타가 바늘구멍에 들어가는 것'으로 비유한다. 미국에서 시판 허가를 받을 가능성은 이보다 더 낮다.
SK바이오팜이 이렇게 작은 구멍을 뚫은 것은 경영진의 신약 뚝심과 장기간 투자가 가능한 자본력이라는 평가가 나온다. 제네릭(복제약) 개발과 생산이 주 수익원이었던 국내 제약 업계에서 신약 개발에 뛰어들어 오랜 기간에 걸쳐 투자했기 때문이다.
최종현 SK 선대 회장은 1993년 대덕연구원에 연구팀을 꾸리면서 신약 개발을 시작했다. 최태원 SK그룹 회장은 바이오 사업에 대한 집념을 이어받았고 결실로 일궈냈다. 2002년 바이오 사업 육성을 목표로 생명과학연구팀, 의약개발 팀 등 나눠져 있던 신약개발 조직을 통합했다. 또, 기술 확보를 위해 중국과 미국에 연구소를 세웠다.
2007년 지주회사 체제로 전환할 때도 신약개발 조직은 분사하지 않고 지주사 직속으로 뒀다. 이후 2011년에는 신약 개발을 집중으로 육성하기 위해 SK바이오팜을 신설했다.
후보물질 발굴부터 임상시험, 허가까지 20년 가까운 기간이 흘렀지만 장기적인 안목으로 적극 투자했기 때문에 가능했다. 업계에서는 회사의 재무 성과가 악화하더라도 수천억 규모의 투자를 단행하는 그룹의 자본력이 신약 개발을 뒷받침했다고 입을 모은다.
업계 관계자는 "SK바이오팜이 당장 재무 성과를 내지 않더라도 그룹 차원에서 시장 성장성과 연구 인력의 전문성을 믿고 장기간 기다려준 것이 성공 비결로 생각된다"라고 했다.
올해 하반기 FDA 승인 국산 신약 나온다… 그 주인공은?

우선 한미약품 롤론티스의 FDA 시판 허가가 오는 9월 결정된다. 처방의약품 신청자 수수료법(PDUFA)으로 정해진 승인 심사 기한은 9월 9일이다. 업계에 따르면 FDA는 최근 한미약품 평택 바이오플랜트를 방문해 실사를 마쳤다. 생산 공장 실사는 품목 허가 절차의 마지막으로 꼽히는 만큼 롤론티스의 미국 승인 가능성도 더 커졌다.
롤론티스는 호중구 감소증 치료제다. 호중구 감소증은 백혈구 촉진제(G-CSF)로도 불리는데 항암 치료 중 감소한 백혈구 수치를 정상화하는 데 사용된다. 호중구 수치가 회복되지 않으면 다음 항암 치료도 지연된다. 이런 의미에서 호중구 감소증 치료제는 항암제는 아니지만 암 치료를 돕는 항암 보조제다.
롤론티스는 2012년 미국 스펙트럼에 기술 이전된 이후 여러 우여곡절을 겪었다. 2018년 스펙트럼이 FDA에 바이오 의약품 품목허가(BLA)를 신청했다가 추가 자료 보완을 이유로 자진 취하했다. 이듬해 재도전했지만 FDA로부터 생산시설 재실사가 필요하다는 보완요구서(CRL)를 받았다. 이후 팬데믹으로 생산시설 재실사가 지연됐고 지난해 8월 다시 CRL을 수령했다. 올해 3월 BLA를 다시 제출했고 네 번째 도전 만에 허가를 목전에 두고 있다.
경쟁 약물로는 뉴라스타가 있다. 호중구 감소증 표준 치료제로 국내외에서 처방 1위를 기록하는 의약품이다. 전 세계 8조원 시장에서 70% 점유율을 차지하는 것으로 알려졌다. 롤론티스에는 한미약품 독자 플랫폼 기술인 '랩스커버리가 적용됐다. 인체 내 의약품 약효 지속 기간을 늘리는 역할을 한다. 앞선 임상 3상 시험에서 롤론티스가 뉴라스타 대비 비열등성을 입증했기에 충분히 경쟁력 있다는 업계 평가가 나온다.
포지오티닙은 미국 시장에 최초로 진출한 국산 항암 신약 타이틀을 노린다. 국내 제약사 항암 신약 중 FDA 승인을 받은 제품은 아직 없다. 포지오티닙은 이전에 치료 경험이 있는 'HER2 Exon 20' 삽입 변이 환자를 위한 비소세포폐암 치료제다. PDUFA에 의한 FDA 승인 심사 기한은 오는 11월 24일이다.
포지오티닙은 2015년 스펙트럼에 기술 이전됐다. 스펙트럼은 'ZENITH20'이라는 임상 2상을 진행 중이다. 총 7개 코호트에서 진행되는데 코호트2 결과를 바탕으로 FDA에 신약 승인을 신청했다. 90명 환자를 대상으로 한 코호트2 연구에서 종양 크기 감소 등을 나타내는 객관적 반응률은 27.8%, 무진행생존기간 중앙값(mPFS)은 5.5개월을 기록하며 FDA로부터 패스트트랙(Fast Track) 지정을 받았다.
비소세포폐암에서 HER2 Exon20 변이 환자 비율은 2% 내외로 알려졌다. 미국에서 한 해 약 3000~4000명 환자가 발생하는 것으로 추정된다. 시장 규모가 크진 않지만 아직 허가된 비소세포폐암 HER2 Exon20 변이 타깃 치료제가 없기에 미충족 의료 수요가 있다.
또한 추가 임상으로 처방 대상을 확보할 여지가 있다. 포지오티닙 코호트4 임상은 이전에 치료 경험이 없는 환자를 대상으로 임상을 진행 중이다. 올해 3월 유럽종양학회 학술대회에서 발표된 결과에 따르면 객관적 반응률이 41%로 집계되는 등 고무적인 기록을 나타냈다. 훨씬 더 많은 사람에게 처방할 수 있는 1차 치료제로 확대될 가능성이 생긴 셈이다.
업계에 따르면 포지오티닙은 내년부터 판매를 시작해 5년 차인 오는 2028년, 약 2억 달러 매출을 올릴 것으로 예상된다.
올해 FDA 신약 승인 문턱을 넘지 못한 기업도 있다. GC녹십자는 아이비글로불린에스엔주(IVIG-SN) 10%에 대해 지난 3월 FDA로부터 CRL을 받았다. FDA가 오창 혈액제제 생산시설을 현장 실사하지 못했다는 게 이유였다.
BLA를 재제출하면 FDA는 현장 실사 후 6개월 이내에 결과를 통보해야 한다. 그러나 GC녹십자의 BLA 제출이 늦어지면서 연내 승인은 어렵게 됐다. GC녹십자 측은 허가와 관련해 FDA와 긴밀히 소통 중이라고만 밝혔다.
지난 3월 메지온의 폰탄수술 환자 치료제 유데나필 FDA 허가도 불발됐다. 임상 3상 1차 평가지표 데이터에서 통계적 유의성을 확보하지 못했기 때문이다.
폰탄수술은 심실을 하나만 갖고 태어난 환자를 대상으로 진행하는 외과 수술이다. 폰탄수술 치료제는 수술 이후 운동 능력을 향상시키는데 아직 전 세계에서 허가받은 약이 없다. 메지온은 추가 임상을 진행해 신약 도전을 계속 이어가겠다고 밝혔다.