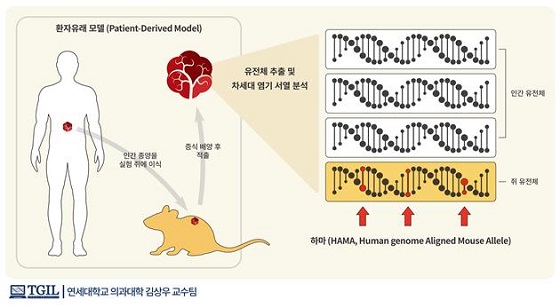
 하마(HAMA)의 정의<br><br>환재유래 모델을 통해 배양된 환자의 암 조직에는 필연적으로 쥐 유전체의 오염이 발생할 수 밖에 없으며 이때 인간 유전서열과의 차이 때문에 검출되는 쥐 유전체 유래의 위양변이를 하마(HAMA, Human genome Aligned Mouse Allele)라고 정의했다/자료=과학기술정보통신부
하마(HAMA)의 정의<br><br>환재유래 모델을 통해 배양된 환자의 암 조직에는 필연적으로 쥐 유전체의 오염이 발생할 수 밖에 없으며 이때 인간 유전서열과의 차이 때문에 검출되는 쥐 유전체 유래의 위양변이를 하마(HAMA, Human genome Aligned Mouse Allele)라고 정의했다/자료=과학기술정보통신부 환자의 치료과정에서 유전자검사, 약물반응검사 등을 위해 종양조직을 여러 차례 분석한다. 이 때문에 한 번 채취한 종양세포를 자연적으로 보존하고 충분히 증식시켜 여러 검사의 시료로 쓸 수 있도록 하는 환자유래모델(PDMS, patient-derived models)이 활용된다.
하지만 이 모델은 종양세포를 주로 생쥐 체내에서 증식시키거나, 생쥐의 세포와 함께 배양하기 때문에 쥐의 세포가 함께 분석돼 자칫 잘못된 결과가 나올 수 있다는 문제점이 있다.
연구팀에 따르면 우선, 쥐와 사람에게서 나타나는 모든 유전자 서열 차이를 찾고, 이를 ‘하마’(HAMA, human-genome aligned mouse allele)라고 명명했다.
특히, 잘 알려진 암 관련 돌연변이 데이터베이스(DB) 정보 중 생쥐를 이용한 실험모델에서 비롯된 경우 유독 ‘하마’의 관찰빈도가 높게 나타나는 것도 확인했다.
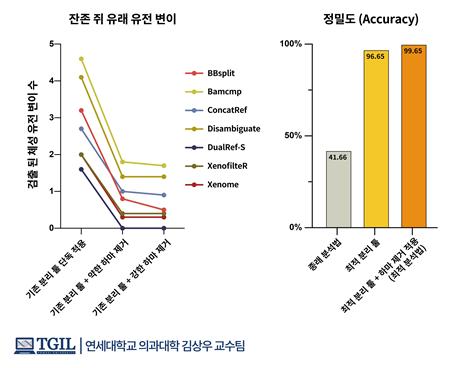 최적 분석법의 성능 분석<br><br>기존에 알려진 쥐 유전체 분리 툴의 성능 분석을 통해 조건별 최적의 성능을 나타내는 분리 툴을 규명 하였으며, 종래 분석법 대비 54.99%의 쥐 유전체 제거 정밀도 향상을 확인하였다. 이 과정에 하마 제거 과정을 추가로 수행할 경우 최종적으로 99.65%의 정밀도로 쥐 유전체 제거가 가능함을 밝혔다/자료=과학기술정보통신부
최적 분석법의 성능 분석<br><br>기존에 알려진 쥐 유전체 분리 툴의 성능 분석을 통해 조건별 최적의 성능을 나타내는 분리 툴을 규명 하였으며, 종래 분석법 대비 54.99%의 쥐 유전체 제거 정밀도 향상을 확인하였다. 이 과정에 하마 제거 과정을 추가로 수행할 경우 최종적으로 99.65%의 정밀도로 쥐 유전체 제거가 가능함을 밝혔다/자료=과학기술정보통신부 아울러 연구팀은 150가지가 넘는 가상의 오염 데이터를 기반으로 비교 분석을 수행해 최적의 오염 배제 방법을 밝혀냈다. 연구팀에 따르면 실제 이를 토대로 최적 유전자분석법을 적용한 결과 기존 분석 대비 정확성을 약 58% 가량 높일 수 있었다.
이 시각 인기 뉴스
김 교수는 “이번 연구는 체외에서 보존, 증식된 환자 암세포 시료의 유전체 분석과정에서 발생할 수 있는 오류를 바로잡는다”며 “앞으로 더 정확한 정보를 통해 환자를 치료할 수 있게 될 것”이라고 말했다. 이번 연구성과는 유전체학 분야 국제학술지 ‘지놈 바이올로지’에 게재됐다.